Data generation pipeline¶
Using the methods below you can generate and update a trackList.json and then make any further manual required.
Canvas vs HTML in Apollo¶
Most of the JBrowse documentation for configuring tracks applies here. However, there are a few important points about Canvas vs HTML tracks.
It should be noted that if you need both benefits of each track type, you are free to duplicate that track and use an alternate styling or track type.
BigWig tracks are only shown as Canvas.
HTML Tracks¶
Pros: Create evidence by dragging and clicking on annotation or evidence does show the alignment. Can use CSS styling. Cons: Renders slower.
Note that in most cases regular HTML rendering will be preferable. Exceptions would be dense BAM alignments and dense Variant tracks where you are attempting to display the density at a higher resolution.
HTML Track mapping with type=<?> :
- Annotation / GFF3:
FeatureTrack,NeatHTMLFeatures/View/Track/NeatFeature,JBrowse/View/Track/HTMLFeatures,WebApollo/View/Track/DraggableNeatHTMLFeatures - Alignment:
JBrowse/View/Track/Alignments,WebApollo/View/DraggableAlignments - Variant:
JBrowse/View/Track/HTMLVariants,WebApollo/View/Track/WebApolloHTMLVariants
Canvas Tracks¶
Pros: Renders faster, non-CSS style options. Cons: Can not drag to create evidence, clicking on annotation or evidence does not show alignment.
Note that in most cases regular HTML rendering will be preferable. Exceptions would be dense BAM alignments and dense Variant tracks where you are attempting to display the density at a higher resolution.
Canvas Track mapping with type=<?> :
- Annotation / GFF3:
NeatCanvasFeatures/View/Track/NeatFeature,JBrowse/View/Track/CanvasFeatures,WebApollo/View/Track/WebApolloNeatCanvasFeatures - Alignment:
JBrowse/View/Track/Alignments2,WebApollo/View/Track/WebApolloAlignments2 - Variant:
JBrowse/View/Track/CanvasVariants,WebApollo/View/Track/WebApolloCanvasVariants
Apollo Automated Configuration and upload¶
Admin users may upload FASTA files to create new genomes and upload most track types in a similar manner if a default configuration is desirable.
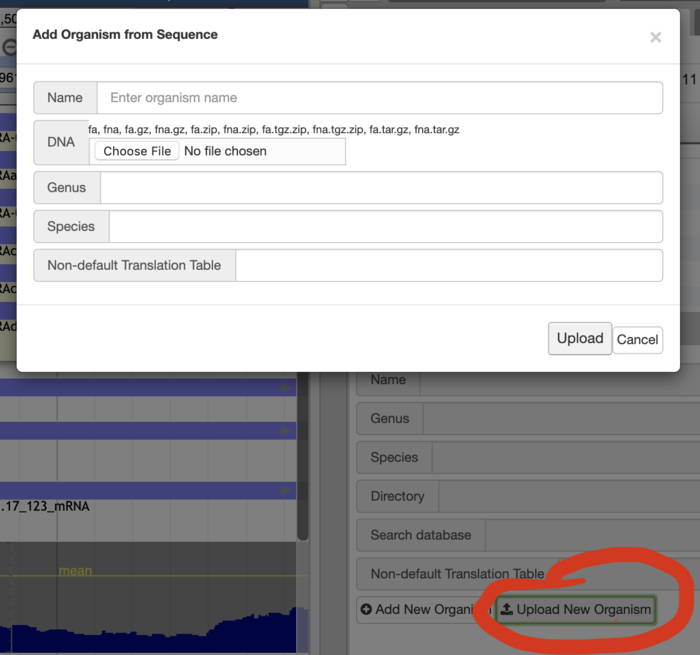
Additionally admin users may also add most tracks in a similar fashion:
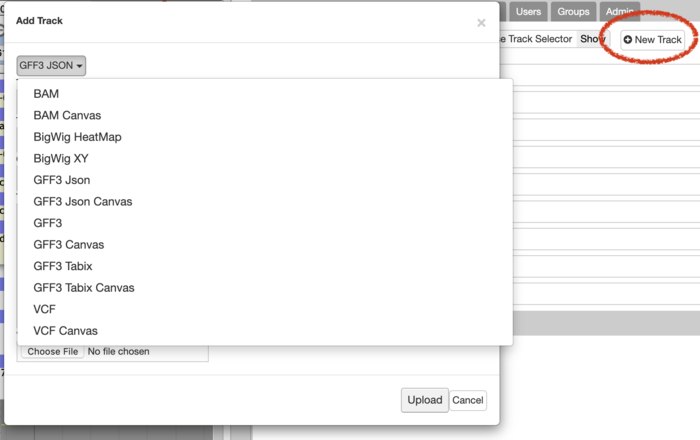
JBrowse Configuration¶
The manual data generation pipeline is based on the typical jbrowse commands such as prepare-refseqs.pl and
flatfile-to-json.pl, and these scripts are automatically copied to a local bin/ directory when you run the setup scripts
(e.g. apollo run-local or apollo deploy or install_jbrowse.sh).
If you don’t see a bin/ subdirectory containing these scripts after running the setup, check setup.log and check the troubleshooting guide for additional tips or feel free to post the error and setup.log on GitHub or the mailing list.
prepare-refseqs.pl¶
The first step to setup the genome browser is to load the reference genome data. We’ll use the prepare-refseqs.pl
script to output to the data directory.
bin/prepare-refseqs.pl --fasta pyu_data/scf1117875582023.fa --out /opt/apollo/data
If you want to use an indexed FASTA genome then you can run prepare-refseqs.pl as follows:
bin/prepare-refseqs.pl --indexed_fasta pyu_data/scf1117875582023.fa --out /opt/apollo/data
The script will copy the genome FASTA and its FAI index into the output folder.
Note: the output directory is used later when we load the organism into the browser with the “Create organism” form
flatfile-to-json.pl¶
The flatfile-to-json.pl script can be used to load GFF3 files and you can customize the feature types. Here, we’ll start off by loading data from the MAKER GFF for the Pythium ultimum data. The simplest loading command specifies a –trackLabel, the –type of feature to load, the –gff file and the –out directory.
bin/flatfile-to-json.pl --gff pyu_data/scf1117875582023.gff --type mRNA \
--trackLabel MAKER --out /opt/apollo/data
Note: you can also use the command bin/maker2jbrowse for loading the MAKER data.
Also see the section Customizing features section for more information on customizing the CSS styles of the Apollo features.
Note: Apollo uses features that are loaded at the “transcript” level. If your GFF3 has “gene” features with “transcript”/”mRNA” child features, make sure that you use the argument –type mRNA or –type transcript.
generate-names.pl¶
Once data tracks have been created, you can generate a searchable index of names using the generate-names.pl script:
bin/generate-names.pl --verbose --out /opt/apollo/data
This is optional but useful step to index of names and features and refseq names. If you have some tracks that have
millions of features, consider only indexing select tracks with the –tracks argument or disabling autocomplete with
--completionLimit 0.
add-bam-track.pl¶
Apollo natively supports BAM files and the file can be read (in chunks) directly from the server with no preprocessing.
To add a BAM track, copy the .bam and .bam.bai files to your data directory, and then use the add-bam-track.pl to add the file to the tracklist.
mkdir /opt/apollo/data/bam
cp pyu_data/simulated-sorted.bam /opt/apollo/data/bam
cp pyu_data/simulated-sorted.bam.bai /opt/apollo/data/bam
bin/add-bam-track.pl --bam_url bam/simulated-sorted.bam \
--label simulated_bam --key "simulated BAM" -i /opt/apollo/data/trackList.json
Note: the bam_url parameter is a URL that is relative to the data directory. It is not a filepath! Also, the .bai will
automatically be located if it is simply the .bam with .bai appended to it.
add-bw-track.pl¶
Apollo also has native support for BigWig files (.bw), so no extra processing of these files is required either.
To use this, copy the BigWig data into the jbrowse data directory and then use the add-bw-track.pl to add the file to the tracklist.
mkdir /opt/apollo/data/bigwig
cp pyu_data/*.bw /opt/apollo/data/bigwig
bin/add-bw-track.pl --bw_url bigwig/simulated-sorted.coverage.bw \
--label simulated_bw --key "simulated BigWig"
Note: the bw_url parameter is a URL that is relative to the data directory. It is not a filepath!
Customizing different annotation types (advanced)¶
To change how the different annotation types look in the “User-created annotation” track, you’ll need to update the
mapping of the annotation type to the appropriate CSS class. This data resides in client/apollo/json/annot.json, which
is a file containing Apollo tracks that is loaded by default. You’ll need to modify the JSON entry whose label is
Annotations. Of particular interest is the alternateClasses element. Let’s look at that default element:
"alternateClasses": {
"pseudogene" : {
"className" : "light-purple-80pct",
"renderClassName" : "gray-center-30pct"
},
"tRNA" : {
"className" : "brightgreen-80pct",
"renderClassName" : "gray-center-30pct"
},
"snRNA" : {
"className" : "brightgreen-80pct",
"renderClassName" : "gray-center-30pct"
},
"snoRNA" : {
"className" : "brightgreen-80pct",
"renderClassName" : "gray-center-30pct"
},
"ncRNA" : {
"className" : "brightgreen-80pct",
"renderClassName" : "gray-center-30pct"
},
"miRNA" : {
"className" : "brightgreen-80pct",
"renderClassName" : "gray-center-30pct"
},
"rRNA" : {
"className" : "brightgreen-80pct",
"renderClassName" : "gray-center-30pct"
},
"repeat_region" : {
"className" : "magenta-80pct"
},
"transposable_element" : {
"className" : "blue-ibeam",
"renderClassName" : "blue-ibeam-render"
}
}
For each annotation type, you can override the default class mapping for both className and renderClassName to use
another CSS class. Check out the Customizing features section for more
information on customizing the CSS classes.
Customizing features¶
The visual appearance of biological features in Apollo (and JBrowse) is handled by CSS stylesheets with HTMLFeatures
tracks. Every feature and subfeature is given a default CSS “class” that matches a default CSS style in a CSS
stylesheet. These styles are are defined in client/apollo/css/track_styles.css and
client/apollo/css/webapollo_track_styles.css. Additional styles are also defined in these files, and can be used by
explicitly specifying them in the –className, –subfeatureClasses, –renderClassname, or –arrowheadClass parameters to
flatfile-to-json.pl (see data loading section).
Apollo differs from JBrowse in some of it’s styling, largely in order to help with feature selection, edge-matching,
and dragging. Apollo by default uses invisible container elements (with style class names like “container-16px”) for
features that have children, so that the children are fully contained within the parent feature. This is paired with
another styled element that gets rendered within the feature but underneath the subfeatures, and is specified by the
--renderClassname argument to flatfile-to-json.pl. Exons are also by default treated as special invisible
containers, which hold styled elements for UTRs and CDS.
It is relatively easy to add other stylesheets that have custom style classes that can be used as parameters to
flatfile-to-json.pl. For example, you can create /opt/apollo/data/custom_track_styles.css which contains two new
styles:
.gold-90pct,
.plus-gold-90pct,
.minus-gold-90pct {
background-color: gold;
height: 90%;
top: 5%;
border: 1px solid gray;
}
.dimgold-60pct,
.plus-dimgold-60pct,
.minus-dimgold-60pct {
background-color: #B39700;
height: 60%;
top: 20%;
}
In this example, two subfeature styles are defined, and the top property is being set to (100%-height)/2 to assure that the subfeatures are centered vertically within their parent feature. When defining new styles for features, it is important to specify rules that apply to plus-stylename and minus-stylename in addition to stylename, as Apollo adds the “plus-” or “minus-” to the class of the feature if the the feature has a strand orientation.
You need to tell Apollo where to find these styles by modifying the JBrowse config or the plugin config, e.g. by adding this to the trackList.json
"css" : "data/custom_track_styles.css"
Then you may use these new styles using –subfeatureClasses, which uses the specified CSS classes for your features in the genome browser, for example:
bin/flatfile-to-json.pl --gff MyFile.gff \
--type mRNA --trackLabel MyTrack \
--subfeatureClasses '{"CDS":"gold-90pct","UTR": "dimgold-60pct"}'
Bulk loading annotations to the user annotation track¶
GFF3¶
You can use the tools/data/add_features_from_gff3_to_annotations.pl script to bulk load GFF3 files with transcripts
to the user annotation track. Let’s say we want to load our maker.gff transcripts.
tools/data/add_features_from_gff3_to_annotations.pl \
-U localhost:8080/Apollo -u web_apollo_admin -p web_apollo_admin \
-i scf1117875582023.gff -t mRNA -o "name of organism"
The default options should be able to handle most GFF3 files that contain genes, transcripts, and exons.
You can still use this script even if the GFF3 file that you are loading does not contain transcripts and exon types.
Let’s say we want to load match and match_part features as transcripts and exons respectively. We’ll use the
blastn.gff file as an example.
tools/data/add_features_from_gff3_to_annotations.pl \
-U localhost:8080/Apollo -u web_apollo_admin -p web_apollo_admin \
-i cf1117875582023.gff -t match -e match_part -o "name of organism"
You can view the add_features_from_gff3_to_annotations.pl help (-h) option for all available options.
Note: Apollo makes a clear distinction between a transcript and an mRNA. Genes that have mRNA as its child feature are treated as protein coding annotations and Genes that have transcript as its child feature are treated as non-coding annotations, specifically a pseudogene.
Note: In order to create meaningful names from your evidence when creating manual annotations, the GFF3 should
provide the Name attribute in column 9 of the GFF3 spec as shown in this example:
NC_000001.11 BestRefSeq gene 11874 14409 . + . ID=gene1;Name=DDX11L1;Dbxref=GeneID:100287102,HGNC:37102;description=DEAD%2FH %28Asp-Glu-Ala-Asp%2FHis%29 box helicase 11 like 1;gbkey=Gene;gene=DDX11L1;pseudo=true
If you would like to look at a compatible representative GFF3, export annotations from Apollo via GFF3 export.
Disable draggable¶
Apollo has a number of specific track config parameters
overrideDraggable (boolean)
determines whether to transform the alignments tracks to draggable alignments
overridePlugins (boolean)
determines whether to transform alignments and sequence tracks
These can be specified on a specific track or in a global config.